
Congenital Cystic Adenomatoid Malformation (CCAM) Treatment
Congenital Cystic Adenomatoid Malformation (CCAM) Treatment
Congenital cystic adenomatoid malformation (CCAM) is a mass of abnormal cysts (bubble balls) lined by proliferating bronchial or cuboidal epithelium with intervening normal portions of lung.
Epidemiology
About 25% of affected infants are stillbirths. 20% may have associated congenital abnormalities. Of the heart lesions, CCAM is most commonly associated with Tetralogy of Fallot (with cyanosis – bluish discolouration).
Of CCAMs diagnosed antenatally, up to 40% may show regression in size of the cysts as the pregnancy progresses.
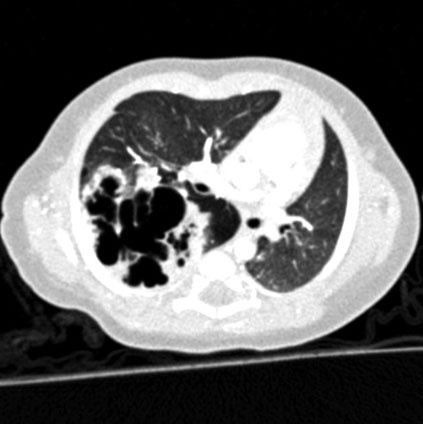
Pathophysiology
It is understood that CCAM occurs when there is failure of interaction between endoderm and mesoderm (primary tissue layers) leading to an imbalance with increased cell proliferation and reduced cell death. This leads to abnormal development of the lung. It is also suggested that CCAM may include sequestration, bronchial atresia and lobar emphysema.
Classification
- Type 0 – acinar atresia
- Type I – cysts up to 10cm. Most common form.
- Type II – Sponge-like multiple small cysts (<2cm) and solid pale tumour-like tissue.Occurs in 40% of patients. Most anomalies (other abnormalities) are associated with type II.
- Type III – Solid. Excess of bronchiolar structures. It is localised to one area
- Type IV – cysts up to 10cm. The cysts are lined by flattened epithelium resting on loose mesenchymal tissue.
Clinical Presentation
CCAMs present in 3 main ways:
- Antenatal diagnosis (symptomatic and asymptomatic): Many resolve progressively on ultrasound.
- Symptomatic post-natal diagnosis.
- Asymptomatic post-natal diagnosis (incidental on CXR).
Signs & Symptoms
- Common symptoms and signs are early tachypnoea and poor feeding.
- Majority of infants are asymptomatic and present later with tachypnoea, repeated chest infections, bronchiectasis and rarely lung abscess.
- Approximately 20% have associated malformations of which the majority are other pulmonary associations. Other associations include renal and cardiac.
- Long term prognosis depends on whether the infant becomes symptomatic with recurrent infections prior to surgical removal.
Investigations
- Often made antenatally, with other possibilities being Congenital Diaphragmatic Hernia (CDH) or sequestrated lung.
- Postnatally the CXR should be done on day 1 of life.
Management
- Infant should be admitted to the neonatal unit and perform an early CXR.
- If the symptoms are related to the CCAM rather than newborn respiratory disease, infant will likely require a CT chest +/- lobectomy if the baby remains symptomatic.
- If asymptomatic, the lesion still needs excision by 1 year of age because of the risk of development of adenomatous tumour.